

پیشبینی ساختار پروتئین به معنای استنتاج ساختار سهبعدی پروتئین از روی دنباله آمینواسیدهای آن یا به بیان دیگر، تعیین ساختار دوم و سوم از روی ساختار اولیهٔ پروتئین است. تعیین ساختار پروتئین از مبنا با مسئلهٔ طراحی یک پروتئین متفاوت است. تعیین ساختار پروتئین یکی از مسائل مهم در حوزه بیوانفورماتیک و شیمی تئوری است و اهمیت زیادی در پزشکی (برای مثال در طراحی دارو) و زیستفناوری (در طراحی آنزیمها) دارد.
اجزای اصلی در ساختار یک پروتئین
پروتئینها زنجیرهای از آمینواسیدها هستند که به وسیلهٔ پیوندهای پپتیدی به یکدیگر متصل شدهاند. شکلهای متفاوتی از این زنجیره (با چرخش در اطراف کربن آلفا) در فضای سهبعدی امکانپذیر است. برخی آمینواسیدها ساختار قطبی دارند و دارای دو ناحیهٔ مجزای مثبت و منفی با یک گروه آزاد C=O و یک گروه آزاد NH هستند. این دو گروه در ساختار پروتئین تشکیل پیوند هیدروژنی میدهند. ۲۰ نوع آمینو اسید موجود را میتواند برمبنای ساختار شیمیایی زنجیره جانبی تقسیمبندی نمود. برای مثال، گلیسین کوچکترین زنجیره جانبی را که تنها شامل یک اتم هیدروژن است، دارد و بنابراین انعطاف بالایی در شکلگیری ساختارهای محلی پروتئین ایجاد میکند.
ساختار پروتئین را میتوان به صورت دنبالهای از اجزای ساختار دوم، حلقههای آلفا و صفحات بتا، در نظر گرفت. در ساختار دوم، الگوهای منظمی از پیوندهای هیدروژنی بین آمینواسیدهای همسایه شکل میگیرد و آمینواسیدها دارای زوایای فی (زاویه حول نیتروژن و کربن آلفا) و سای (زاویه حول پیوند کربن آلفا و کربن کربونیل) یکسانی هستند.
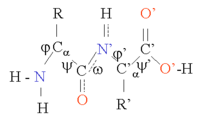
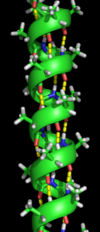
مارپیچ آلفا
مارپیچ آلفا پر تکرارترین نوع از اجزای ساختار دوم در پروتئینها است که در هر دور بهطور متوسط ۳٫۶ آمینواسید دارد. یک پیوند هیدروژنی نیز در انتهای دور چهارم ایجاد میشود. طول متوسط هر مارپیچ ۳ دور (معادل با ۱۰ آمینواسید) است. این طول از ۱٫۵ تا ۱۱ دور (۵ تا ۴۰ آمینواسید) متغیر است. همترازی پیوندهای هیدروژنی یک گشتاور دوقطبی را در مارپیچ ایجاد میکند و منجر به ایجاد یک بار مثبت جزئی در آمینواسید انتهای مارپیچ میشود. از آنجا که این ناحیه دارای گروه آزاد NH است، با یک گروه با بار منفی مانند فسفات تعامل دارد. معمولترین مکان برای مارپیچ آلفا سطح هستههای پروتئین است که یک رابط را برای تعامل با محیط آبی بیرون ایجاد میکند. قسمت داخلی مارپیچ تمایل به داشتن آمینواسیدهای آبگریز و قسمت بیرون تمایل به داشتن آمینواسیدهای آبدوست دارد؛ بنابراین در طول مارپیچ از هر چهار آمینواسید، سه آمینواسید آبگریز خواهند بود. سایر آمینواسیدهای موجود در هسته پروتئین یا داخل غشای سلولی خاصیت آبگریزی دارند. بهطور کلی مارپیچهای قرار گرفته در سطح، تعداد کمتری آمینواسید آبگریز دارند. از این ویژگی میتوان در پیشبینی ساختار پروتئینها کمک گرفت. برای مثال نواحی با مقادیر بیشتر از آلانین، گلوتامین اسید، لوسین و متیونین و مقادیر کمتر از پرولین، گلیسین، تیروزین و سرین تمایل به تشکیل مارپیچ آلفا دارند.
صفحات بتا
صفحات بتا از پیوندهای هیدروژنی میان ۵ تا ۱۰ آمینواسید متوالی در یک بخش از زنجیره با ۵ تا ۱۰ آمینواسید دیگر در ناحیهای دیگر از زنجیره تشکیل شدهاند. این نواحی دارای کنش شیمیایی ممکن است همسایه باشند (با یک حلقهٔ کوتاه در میانشان) یا ممکن است خیلی دور باشند و ساختارهای متفاوتی در میانشان وجود داشته باشد. زنجیرهها در صفحه میتوانند همجهت باشند که تشکیل یک صفحه موازی را میدهند، همچنین میتوانند در جهت متفاوت باشند که صفحهٔ ناموازی نامیده میشوند و در نهایت یک صفحه میتواند هر دو دسته زنجیره را داشته باشد که صفحه ترکیبی نام دارد. الگوی پیوندهای هیدروژنی در صفحات موازی و ناموازی متفاوت است. هر آمینواسید در رشتهٔ داخلی صفحه دو پیوند هیدروژنی را با آمینواسیدهای همسایه تشکیل میدهد، در حالی که آمینواسیدهای رشتههای مرزی تنها یک پیوند هیدروژنی را با رشتهٔ مجاور داخلی میدهند. بهطور کلی، پیشبینی مکان صفحات بتا از مارپیچهای آلفا مشکلتر است.
حلقه
حلقهها نواحی از زنجیره پروتئینی هستند که (۱) بین مارپیچهای آلفا و صفحات بتا هستند، (۲) دارای طولهای متفاوت و ساختار سهبعدی هستند و (۳) در سطح ساختار قرار دارند. حلقههای با پیچ تند که یک دور کامل در زنجیره پلیپپتیدی هستند و دو رشته ناموازی بتا را به هم متصل میکنند، ممکن است طولی به اندازهٔ دو آمینواسید داشته باشند. حلقهها با محیط آبی اطراف و سایر پروتئینها در تعاملاند. از آنجا که آمینواسیدهای روی حلقه همانند آمینواسیدهای داخل هسته محدودیت فضا و محیط ندارند و همچنین تأثیری روی شکلدهی به ساختار دوم ندارند، احتمال رخداد جایگزینی، حذف یا جایگذاری آمینواسیدها در آنها بیشتر است؛ بنابراین، در یک دنباله همترازی، وقوع بیشتر این موارد میتواند نشاندهندهٔ یک حلقه باشد. نقش حلقهها در دنبالهٔ پروتئینی همانند نقش نواحی اینترون در دنباله ژنوم است.
چنبره
ناحیهای از ساختار دوم که یک مارپیچ آلفا یا یک صفحه بتا نباشد و نتوان آن را یک حلقه در نظر گرفت، به عنوان یک چنبره در نظر گرفته میشود.
ساختار دوم
پیشبینی ساختار دوم به مجموعه روشهایی در بیوانفورماتیک گفته میشود که به تعیین ساختار محلی دوم از روی دنباله آمینواسیدها میپردازند. تعیین ساختار دوم به معنای تعیین مکان مارپیچهای آلفا، صفحات بتا و حلقههای روی رشته آمینواسید است. میزان موفقیت یک روش تعیین ساختار دوم را میتوان به کمک نتایج به دست آمده از الگوریتم DSSP روی ساختار بلور پروتئین ارزیابی نمود. بهترین و جدیدترین روشهای موجود در این حوزه دقتی حدود ۸۰ درصد دارند. این دقت بالا موجب میشود تا از ساختار پیشبینی شده بتوان به عنوان ویژگی در بهبود مسائلی نظیر طبقهبندی الگوهای ساختاری یا همترازی دنبالههای پروتئینی استفاده نمود. دقت روشهای تعیین ساختار دوم، به صورت هفتگی توسط LiveBench و EVA تعیین میشود.
مروری بر الگوریتمهای پیشنهادی
امروزه، بیش از ۲۰ روش متفاوت برای پیشبینی ساختار دوم وجود دارد. یکی از اولین روشها، روش کو-فاسمن است که بر مبنای احتمال رخداد هر یک از آمینواسیدها در هر ساختار عمل میکند. پارامترهای مربوط به هر احتمال بر مبنای تعداد رخداد هر آمینواسید در هر یک از اجزای ساختار دوم به دست میآید. اولین بار در اواسط دهه ۱۹۷۰ میلادی، نتایج ضعیفی از این الگوریتم به کمک پارامترهای به دست آمده از یک نمونه کوچک از دنبالههای آمینواسیدی به دست آمد. درسالهای بعد روشهای جدیدتری برای تخمین بهتر پارامترهای هر احتمال ارائه شد. دقت این روش در اطراف ۵۰ تا ۶۰ درصد است.
الگوریتم قابل توجه بعدی در این حوزه روش GOR (برگرفته از نام سه دانشمندی که در این حوزه کار میکردند) است که بر مبنای تئوری اطلاعات کار میکند. این روش نسبت به روش قبلی از تکنیک احتمالاتی قویتر استنتاج بیزی استفاده میکند. این تکنیک علاوه بر احتمال رخداد هر آمینواسید در هر ساختار از احتمال شرطی رخداد هر آمینواسید به شرط همسایههایش نیز استفاده میکند. این روش نسبت به الگوریتم کو-فاسمن دقیقتر و حساستر است. دقت اولین نوع از الگوریتم GOR برابر با ۶۵ درصد است. این روش مارپیچهای آلفا را بهتر از صفحات بتا تشخیص میدهد.
گام رو به جلوی بعدی در این حوزه، بهکارگیری روشهای یادگیری ماشین همچون شبکههای عصبی مصنوعی است. مجموعهای از ساختارهای تعیین شده روی یک سری دنباله را به هر شبکه عصبی مصنوعی به عنوان داده آموزش داده میشود تا شبکه، الگوهای مشترک در هر ساختار را شناسایی کند. این دسته از روشها دارای دقت بیش از ۷۰ درصد هستند. با این وجود، صفحات بتا همچنان با دقت بسیار پایینی تعیین میشوند. این نکته به دلیل کمبود اطلاعات در رابطه با ساختار سهبعدی و الگوهای پیوندهای هیدروژنی است. دو روش شناخته شده بر مبنای شبکههای عصبی "PSIPRED" و "JPRED" هستند. در پژوهشهای بعدی، ماشین بردار پشتیبان نیز به عنوان روشی کارآمد برای یافتن حلقهها معرفی شد.
ساختار سوم
دو زیر مسئلهٔ مهم در تعیین ساختار پروتئین، محاسبهٔ انرژی آزاد آن و یافتن کمینه سراسری این انرژی است. یک الگوریتم پیشبینی ساختار پروتئین باید تمام فضای ساختارهای ممکن یک پروتئین را برای یافتن کمینه سراسری جستجو کند. ابعاد این فضا بسیار بزرگ است. یک رویکرد برای کاهش فضای جستجو هرس کردن آن است. میتوان از ساختارهایی که به صورت آزمایشگاهی از پروتئینهای مشابه پروتئین هدف تعیین شدهاند، برای هرس کردن فضای جستجو، استفاده کرد. زانگ در مقالهٔ خود پیشرفتها و چالشهای پیش روی روشهای تعیین ساختار پروتئین را مطرح کردهاست.
روشهای مبتنی بر انرژی
روشهای تصادفی زیادی برای جستجوی فضای ساختارهای پروتئین برای یافتن کمینه سراسری ارائه شدهاست. این دسته از روشها منابع محاسباتی بالایی نیاز دارند و تنها برای پروتئینهای کوچک کارآمد هستند. برای پروتئینهای بزرگتر نیاز به گسترش الگوریتمهای بهینه و همچنین منابع محاسباتی عظیمی همچون ابرکامپیوترها یا محاسبات توزیع شده است.
مدلسازی مقایسهای پروتئین
مدلسازی مقایسهای پروتئین، ساختارهای معین پروتئینهای دیگر را به عنوان یک نقطه شروع یا الگو استفاده میکند. این روش به این دلیل کارآمد است که با وجود تعداد زیادی پروتئین تنها تعداد محدودی ساختار سهبعدی ممکن برای پروتئینها وجود دارد. تخمین زده میشود که با وجود میلیونها نوع پروتئین تنها حدود ۲۰۰۰ نوع تاشدگی متفاوت در فضای سه بعدی برای پروتئینها وجود دارد. این روشها را میتوان در دو گروه زیر قرار داد:
مدلسازی همولوژِیکی
این دسته از روشها برمبنای این فرض منطقی هستند که دو پروتئین با دنباله توالی مشابه دارای ساختارهای سه بعدی مشابهی نیز هستند. دقت این دسته از روشها به میزان تشابه بین پروتئین مورد نظر و پروتئینهای مورد استفاده الگوریتم بستگی دارد. مشکل عمده مدلسازی همولوژیکی پیدا کردن توالی هدفی است که به عنوان الگو استفاده میشود. از آنجا که تاشدگیهای پروتئین در طول روند تکامل کمتر از خود توالی دچار تغییر میشوند، با دقت مناسبی میتوان از توالیهایی که حتی در همترازی فاصلهٔ زیادی دارند، به عنوان الگو استفاده نمود.
مدلسازی بر اساس رشته پروتئین
در این روش رشتهٔ پروتئین با مجموعه پروتئینهای با ساختار مشخص در یک پایگاه داده مقایسه میشود. برای هر یک به کمک یک تابع امتیازدهی، میزان سازگاری هر دنباله با هر ساختار مشخص میشود؛ بنابراین ساختارهای ممکن برای هر دنباله معین میشوند.
پیشبینی هندسه زنجیره جانبی
قرارگیری زنجیرهٔ جانبی آمینواسیدها در داخل ساختار سه بعدی نیز یکی از مسائل مطرح در پیشبینی ساختار پروتئینها است. این مسئله معمولاً به صورت قرارگیری مجموعهای از پیکربندی زنجیرهها بر روی یک ستون محکم پلی پپتیدی مطرح میشود. به این مجموعه پیکربندیها "رتیمر" گفته میشود. این دسته از الگوریتمها به دنبال یافتن مجموعهای از این پیکربندیها هستند که انرژی تمام مجموعه کمینه شود. دو رویکرد مطرح در این حوزه الگوریتمهای "DEE" و "SCMF" هستند که یک تابع هزینه را روی یک سری متغیر گسسته کمینه میکنند که در اینجا این متغیر گسسته رتیمرها هستند. این الگوریتمها از کتابخانههای موجود رتیمرها استفاده میکنند. این کتابخانهها در هر یک از سه دسته مستقل از ستون پروتئین، وابسته به ستون پروتئین یا وابسته به ساختار دوم قرار میگیرند. کتابخانههای مستقل از ستون پروتئین هیچگونه اطلاعاتی از پیکربندی خود ستون نمیدهند. کتابخانههای وابسته به ساختار دوم، آمارهای مربوط به هر نوع رتیمر را برای هر یک از اجزای ساختار دوم یعنی مارپیچ آلفا، صفحه بتا و حلقهها به صورت مجزا ارائه میدهند. در نهایت، کتابخانههای وابسته به ستون پروتئین نیز آمارهایی وابسته به پیکربندیهای محلی ستون در نقاط مختلف ارائه میدهند.
ساختار چهارم
درشرایطی که توانسته باشیم ساختار سهبعدی هر پروتئین را با دقت بالایی پیشبینی کنیم، میتوان در مورد ساختار چهارم که مربوط به قرارگیری دو یا چند پروتئین در کنار هم است نیز اظهار نظر نمود.
نرمافزارها و ابزارهای محاسباتی
امروزه، تعداد زیادی نرمافزار برای برای تعیین ساختار پروتئینها وجود دارد. دو ابزار موفق در این زمینه "I-TASSER" و"HHpred" نام دارند.