

نشانگان سرطانی یا نشانگان سرطان خانوادگی، یک آشفتگی و اختلال ژنتیکی است در آن کسی که جهشهای ژنی در یک یا بیشتر ژنهای تکی مستعدی که تغییر یافتهاند برای پیشروی و پیشرفت سرطان را به ارث برده و میتواند منجر به تاخت و تاز و هجوم دیگر سرطانها نیز شود. نشانگانهای سرطان معمولاً بهتنهایی یک خطر زندگی بزرگ پیشروی سرطان ظاهر نمیشوند، ولی پیشرفت چندگانه وابسته به تومورهای نخستین دانسته میشوند. بسیاری از این نشانگانها بهواسطهٔ جهشهایی در ژنهای سرکوبگر تومور رخ میدهد که این ژنها در فراهم آوری سلولهای توموری سرطاندار نقش دارند. دیگر ژنهایی که میتوانند در تعمیر دیانای مؤثر باشند؛ آنکوژنها و ژنهایی هستند که در ساخت رگهای خونی (رگزایی) شرکت میکنند. نمونههای رایج از نشانگانهای سرطان ارثی، نشانگان سرطان پستان – تخمدان و سرطان روده بزرگ بی پولیپوزیز ارثی (نشانگان لینچ) میباشند.
پیشزمینه
نشانگانهای سرطان ارثی زیر ۵٪ تا ۱۰٪ همهٔ سرطانها قرار دارند. دانستن علمی یک سرطان؛ آمادگی نشانگانهای که بهطور فعال بسط یافتهاند: نشانگانهای زاید و اضافی یافت شدند، زیستشناسی، اساساً واضحتر شد و تجاریسازی روششناسی ژنتیک تشخیصی در بهبودی بیماریهای بالینی انجام یافت. شیوع سرطانّهای پستان و رودهٔ بزرگ که بیشترین نشانگانهای بازشناختی را داراست و شامل نشانگان سرطان تخمدان – پستان ارثی و سرطان روده بزرگ نا-پولیپوزیز ارثی (نشانگان لینچ) میباشد.
برخی سرطانهای کمیاب و نادر با نشانگانهای آمادگیکنندهٔ سرطان ارثی قویا درآمیختهاند. آزمونهای ژنی شاید بتوانند با کارسینومای فوق کلیوی با تومورهای سرطانواره؛ سرطان لیومیوسارکوم؛ سرطان مغزینهای تیروئید؛ سرطان پاراگانگلیون یا فئوکروموسیتوما؛ کارسینومای سلول کلیوی رنگگریز؛ آنکوسیتیک دورگهی یا بافتشناسی سرطان آنکوسیتی؛ کارسینومای چربی؛ و تومورهای طناب جنسی با لولههای حلقوی.
دانش ژنتیک

دو نسخهٔ از هر ژنی که در تمام یاختههای بدن هر کسی حضور دارد را «آلل» مینامند. بیشتر نشانگانهای سرطان در یک راه مندلی اتوزومی غالب پخش میشود. در این موارد، تنها یک آلل معیوب میتوان بهتنهایی سرطان را در شخص ایجاد نماید. کسانی که دارای چنین آللی هستند را «هتروزیگوس» و کسانی که دارای دو آلل یکسان چه معیوب-معیوب و چه سالم-سالم را «هموزیگوس» خوانند. برپایه ژنتیک مندلی از آمیزش یک هتروزیگوس با یک هموزیگوس، زادههای حاصل ۵۰٪ احتمالاً دارای آن بیماری هستند. جهش در ژن به ارث برده شده بانام «یک جهش جنینی» شناخته میشود و یک جهش بیشتر در یک آلل طبیعی در پیشرفت و توسعهٔ سرطان نتیجه میدهد. این رخداد بانام «فرضیهٔ دو ضربهای کنودسون» شناخته میشود که نخستین ضربه به ژن یک ژن ارثی است و دومین در زندگی رخ میدهد. همانگونه که یک آلل نیازمند این است که جهش یافته شود (قیاسا هر دوی آنها را «سرطانهای پراکنده» میخوانند)، احتمال جهش تکی بیشتر از احتمال دو جهش پیاپی در جمعیت است.
کمترین احتمال، نشانگانهایی هستند که به صورت آتوزومی بازگشتی در دودمانه پخش میشوند. جفت آللهای ژن بایستی جهشیافته باشد در اختلالات بازگشتی آتوزومی برای آمادهسازی آن به سرطان است. کسی که با دو آلل بازگشتی است به عنوان یک «هموزیگوس بازگشتی» دانسته میشود. هر دوی والدین بایستی یک آلل معیوب و یک طبیعی داشته باشند تا زادهای هموزیگوس بازگشتی پدیدآورند. اگر والدین دارای یک آلل جهشیافته و یک آلل طبیعی (هتروزیگوس) باشند، ۲۵٪ احتمال زادهٔ هوموزیگوس بازگشتی، ۲۵٪ احتمال زادهٔ هتروزیگوس که حامل ژن معیوب است و ۲۵٪ احتمال زادهٔ هوموزیگوس سالم و طبیعی را دارند.
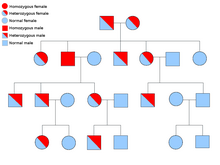
نمونههای نشانگانهای سرطان آتوزومی غالب: نشانگان خودایمنی تکثیر لمفوسیتی که بانام نشانگان کانل – اسمیت شناخته میشود؛ نشانگان بکواید – ویدمان که ۸۵٪ موارد سرطانهای پراکنده هستند؛ نشانگان بیرت – هوگ – دوبه؛ نشانگان کانری؛ نشانگان کودِن؛ سرطان نخاع خانوادگی؛ نشانگان خال دیسپلازی با سرطان ملانین، پولیپوزیس غدهای خانوادگی؛ نشانگان سرطان پستان – تخمدان ارثی؛ سرطان معدی پخششوندهٔ ارثی؛ سرطان رودهبزرگ نا پولیپوزیس ارثی یا نشانگان لینچ؛ نشانگان لی – فرومنی؛ نئوپلازیای درونریز چندگانهٔ تیپ ۲/۱؛ استئوکندروماتوزیز چندگانه؛ نشانگان پوتز – جگرز؛ سرطان پروستات خانوادگی؛ سرطان یاختهٔ کلیوی لیومیوماتوزیز ارثی؛ سرطان سلول کلیوی برآمدگی ارثی؛ نشانگان پاراگانگلیوما – فنوکروموسیتوما ارثی؛ تومور شبکیه؛ تصلب تکمهای بیماری فون هیپل – لینداو و تومور ویلم.
نمونههای نشانگانهای سرطانی بازگشتی آتوزومی: آتاکسی تلانژیکتازی، نشانگان بلوم، کمخونی فانکُنی، پلیپوزیس همبسته با MUTYH، نشانگان روتموند – تامسون، نشانگان ورنر و خشکپوستی رنگدانهای.
برخی نمونهها
گرچه نشانگانهای سرطانی یک خطر افزایش یافته سرطان را نمایش میدهند ولیکن خطر متفاوت است. برای برخی از آن بیماریها، سرطان در آغاز بروز نمیکند. در اینجا بحث بر روی خطر افزایشی سرطان همراه آن متمرکز شدهاست. این سیاهه به دور از پرچانگی و توضیح زیاد است.
کمخونی فانکنی
کمخونی فانکنی (FA) یک اختلال همراه با طیفی پهناور نشانههای بالینی است که شامل: آغاز زودرس و افزایش خطر سرطان، نقصان مغز استخوان و ناهنجاریهای مادرزادی میباشد. برجستهترین ظهور این اختلال وابستگیاش به خونسازی تولید خون به واسطهٔ مغز استخوان) بوده که دربردارندهٔ: کمخونی آپلاستیک، نشانگان میلودیزپلاستیک و لِکومیا میلوئید حاد است. تومورهای کبدی و سرطان سلولهای سنگفرشی مری، گلو و زبان کوچک معمولاً تومورهای سخت و سفت در ارتباط با کمخونی فانکنی هستند. ناهنجاریهای مادرزادی شامل: ناهنجاریهای استخوانی (به ویژه بر روی دستها اثر میکند)، خالهای کافه آو لایت و هیپوپیگمانتاسیون هستند. امروزه، ژنهای شناخته شدهای که منجر به کمخونی فانکنی میشوند، عبارتاند از: FANCA, FANCB, FANCC, FANCD2، FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP و BRCA2 (پیشتر با نام FANCD1 شناخته میشد). ارث این نشانگان عمدتاً آتوزومی بازگشتی است ولی FANCB میتواند از کروموزومهای x پدری یا مادری (ارثیت بازگشتی x-پیوندی) به ارث برسد. مسیر کمخونی فانکنی در تعمیر DNA درگیر میشود، هنگامی که دو رشتهٔ DNA به صورت نادرست بهم الحاق میشوند (کراس لینکهای درونرشتهای). بسیاری مسیرها با مسیر کمخونی فانکنی هماهنگ میشوند از اینرو شامل تعمیر بُرِش نوکلئوتید، سنتز ترجمه و نوترکیبی همتا میباشند،،،،،
پولیپوزیس غدهای خانوادگی
پولیپوزیس غدهای خانوادگی (FAP) یک نشانگان غالب آتوزومی است که خطر سرطان روده-مقعدی را بهطور زیادی افزایش میدهد. از هر ۸۰۰۰ تن، یکی این بیماری را داشته و این بیماری تقریباً نافذیت ۱۰۰٪ دارد. فرد دچار بیماری در رودهٔ بزرگ خود صدها هزار از این غدههای سرطانی را خواهد داشت و در بیشتر موارد به سرطان مترقی و پیشرونده تغییر میکند. دیگر تومورهای افزایش یافته در فراوانی شامل: سرطانهای استخوانی، سرطانها و کارسینوماهای غدههای فوق کلیوی، تومورهای تیروئید و تومورهای دسموئید هستند. عامل این اختلال یک ژن APC جهشیافتهاست که در تنظیمβ-catenin شرکت میکند. APC معیوب منجر به انباشتن β-catenin در یاختهها و فعالسازی عوامل رونویسی درگیر در تکثیر، مهاجرت، تمایز و مرگ برنامهریزی شده سلولی یا آپوپتوز میشود،،.
سرطان تخمدان و پستان ارثی
نشانگان سرطان پستان – تخمدان ارثی (HBOC) یک اختلال ژنی آتوزومی غالب که با جهشهای ژنی بر ژنهای BRCA1 و BRCA2 رخ میدهد. در زنان این اختلال عمدتاً خطر سرطان پستان و تخمدان را افزایش میدهد ولی خطر کارسینومای لولهٔ فالوپ و کارسینومای خونابهدار برآمدهٔ صفاق را نیز افزایش میدهد. در مردان خطر سرطان پروستات افزایش مییابد. دیگر سرطانهایی که متناقضانه با این نشانگان پیوند خوردهاند؛ سرطان لوزالمعده، سرطان پستان نرینه، سرطان روده بزرگ – مقعدی و سرطانهای گردن رحم و رحم. جهشهای ژنی تقریباً ۷٪ و ۱۴٪ سرطان پستان و تخمدان، به ترتیب، BRCA1 و BRCA2، ۸۰٪ موارد را تخمین میزنند. BRCA1 و BRCA2 هر جفت ژنهای سرکوبگر تومور مستلزم شده در بقا و تعمیر DNA هستند. جهشها بر روی این ژنها اجازه کمک به تخریب DNA را داده که میتواند منجر به سرطان شود،
سرطان رودهٔ بزرگ نا-پولیپوزیس ارثی
سرطان رودهٔ بزرگ نا-پولیپوزیس ارثی (HNPCC) که بانام نشانگان لینچ نیز شناخته میشود، یک نشانگان سرطان غالب آتوزومی است که خطر سرطان روده بزرگ – مقعدی را افزایش میدهد. این سرطان با جهشهایی ژنی در تعمیر ناجور DNAیا MMR رخ میدهد، ژنهای شایان توجه MLH1، MSH2، MSH6 و PMS2 هستند. افزون بر سرطان رودهٔ بزرگ – مقعدی، فراوانی بسیاری سرطانهای دیگر نیز افزایش مییابد. آنها عبارت اند از: سرطان اندوتلیال، سرطان معده، سرطان تخمدان، سرطانهای رودهٔ کوچک و سرطان لوزالمعده. همچنین HNPCC با یک هجوم آغازین سرطان رودهٔ بزرگ – مقعدی همراه شدهاست. ژنهای MMR در تعمیر DNA هنگامی که بازها بر روی هر رشته از DNA جور نشود، درگیر میشوند. ژنهای MMR معیوب اجازه الحاق ممتد و جهشهای حذف در مناطقی از DNA که با عنوان ریزماهوارهها شناخته میشود را میدهد. این توالیهای کوتاه تکراری DNA ناپایدار گشته، منجر به یک حالت نااستواری ریزماهواره میشود. ریزماهوارههای جهشیافته معمولاً در ژنهای درگیر در راهاندازی و پیشرفت تومور یافت میشوند و MSI میتواند ابقای زیستی یاختهها را بالا برده و منجر به سرطان گردد،،

نشانگان پاراگانگلیونوما – فنوکروموسیتومای ارثی
بیشتر موارد پاراگانگلیونومای خانوادگی با جهشهایی در ژنهای (SDHD, SDHAF2, SDHC, SDHB) زیرواحد آنزیم سوکسینات دهیدروژناز یا SDH (succinate :ubiquinone oxidoreductase) رخ میدهد. جهش SDHDهمراه با PGL-1 همبسته شده و اکثراً PGL-1 به تنهایی با پاراگانگلیونومای پدری بیشتر از مادری مؤثرتر است. PGL1 و PGL2 با هم دیگر غالب همبارز هستند. PGL-4 با جهش SDHB همبسته شد و خطر بیشتر فنوکروموسیتوما و به همین روال سرطان سلول کلیوی و سرطان تیروئید نا-مغزینهای (non-medullary thyroid cancer) را بالا میبرد.
نشانگان لی – فرومنی
نشانگان لی – فرومنی یک سندرم غالب آتوزومی است که معمولاً با جهشهایی در ژن TP53 رخ میدهد، که منجر به افزایش زیاد خطر سرطانهای بسیاری و نیز شروع زودتر همراه با سرطان میشود. سرطانهای در پیوند با این اختلال شامل: سارکوماهای بافتی نرم (در کودکی بیشتر یافت میشود)، اوسئوسارکوما، سرطان پستان، سرطان مغز، لِکِمیا (leukaemia) و کارسینومای قشر فوق کلیوی (adrenocortical carcinoma). افراد با نشانگان لی – فرومنی معمولاً دارای سرطانهای ابتدایی ناوابستهٔ چندگانه هستند. دلیل برای این طیف بزرگ بالینی از اختلالات شاید به خاطر جهشهای ژنی باشد که بیماری را تعدیل میکند. فراوردهٔ پروتئینی که با ژن TP53 ساخته شده، p53، در بازدارندگی چرخهٔ یاخته، تعمیر DNA و آپوپتوز درگیر است. p53 معیوب میتواند برای ایفای خالصانهٔ پردازشها توانا نباشد که شاید علتی برای تشکیل تومور باشد. زیرا به تنهایی ۶۰–۸۰٪ افراد دچار اختلال دارای جهشهای یافتنی در TP53 است، دیگر جهشها در مسیر p53 شاید در نشانگان لی – فرومنی شرکت کنند.
پولیپوزیس همبسته شده با MUTYH
پولیپوزیس همبسته شده با MUTYH بیشترین جزئیات بالینی را با FAP به اشتراک میگذارد که تمایزی با یک اختلال آتوزومی بازگشتی را با جهشهایی درMUTYH ژن تعمیر DNA منجر میشود. تومورهای افزایش خطری در این اختلال، سرطان رودهٔ بزرگ – مقعدی، آدنوماهای معدی و آدنوماهای دوازدههای هستند.
نشانگان کارسینومای سلول پایهای خالدار
نشانگان کارسینومای سلول پایهای خالدار (NBCCS) که همچنین با نام نشانگان گورلین (Gorlin syndrome) شناخته میشود، یک نشانگان سرطان غالب آتوزومی است که در هنگامی که کارسینومای سلول پایهای خطرش بسیار بالا باشد، رخ میدهد. بیماری با سلول خالدار پایهای، کراتوسیتها و ناهنجاریهای اسکلتی تشخیص داده میشود. تخمینهای شیوع NBCCS متفاوت بوده ولی تقریباً ۱ در ۶۰۰۰۰ است. حضور کارسینومای سلول پایهای در افراد سفیدپوست نسبت به سیاهپوست بسیار بالاتر بوده؛ به ترتیب ۸۰٪ و ۳۸٪ است. کراتوسیتهای دندانزایی (Odontogenic keratocysts) تقریباً در ۷۵٪ افراد دچار این بیماری یافت میشود و معمولاً در آغاز زندگی رخ میدهد. بیشترین ناهنجاریهای اسکلتی رایج در سر و چهره اتفاق میافتند ولی در دیگر مناطق گاهی مانند قفسه سینه نیز کارگر میشود. جهش ژنی مؤثرهٔ بیماری در ژن PTCH رخ میدهد و فراوردهٔ PTCH یک سرکوبگر تومور بوده که در پیامدهی سلول شرکت میکند. گرچه نقش صحیح این پروتئین در NBCCS ناشناخته است، ولی آن در مسیر پیامدهی جوجه تیغی (hedgehog signaling pathway) که در کنترل رشد و توسعه سلول شناخته شده، درگیر است.
بیماری فون هیپل – لینداو

بیماری فون هیپل – لینداو (VHL) یک حالت نادر، ژنی غالب آتوزومی است که افراد دچار را مستعد به تومورهای بدخیم و خوشخیم میکند. بیشتر تومورهای رایج در VHL دستگاه عصبی مرکزی و همانژیوبلاستوماهای شبکیهای (retinal hemangioblastomas)، کارسینوماهای کلیوی سلولی تمیز (clear cell renal carcinomas)، فنوکروموسیتوماها، توموردارهای عصب-درونریز لوزالمعدی (pancreatic neuroendocrine tumours)، کیستهای لوزالمعدی، تومورهای کیسهٔ درون لمفاوی (endolymphatic sac tumors) و کیستادنوماهای نوکدار اپیدیدیمی. (epididymal papillary cystadenomas) VHL از یک جهش در ژن سرکوبگر تومور فون هیپل – لینداو بر کروموزوم 3p25.۳ ناشی میشود.
خشکپوستی رنگدانهدار
خشکپوستی رنگدانهدار (XP) یک اختلال بازگشتی آتوزومی است که با حساسیت بر پرتوی فرابنفش تشخیص داده میشود، خطر بزرگ افزایش آفتاب سوختگی و خطر افزایش سرطانهای پوست را منجر میشود. خطر سرطان پوست بیشتر از ۱۰۰۰ بار نسبت به افراد طبیعی است و شامل تیپهای سرطان پوست بسیاری همچون سرطانهای پوست ملانوما (melanoma) و نا-ملانوما (non-melanoma) است. همچنین، مناطق در معرض آفتاب، زبان، لبها و چشمها خطری افزایش یافتهای از سرطانی شدن را دارند. XP شاید با دیگر سرطانهای درونی و تومورهای خوشخیم همراه باشد. افزون بر سرطان، برخی جهشهای ژنی که منجر به XP میشوند با عصب-تباهی (neurodegeneration) همراه هستند. XP میتواند با جهشهای ژنی در ۸ ژن که پیرو آن فراوردههایی آنزیمی دارند، عبارت اند از: XPA, XPB, XPC, XPD, XPE, XPF, XPG و Pol η. XPA-XPF، آنزیمهای تعمیر برشی نوکلئوتید هستند که DNA آسیب دیده از پرتوی فرابنفش را تعمیر میکند و اجازهٔ ساخت جهشهای پروتئینهای معیوب را به واسطهٔ پرتوی فرابنفش میدهد. Pol η یک پلیمراز بوده که یک آنزیم درگیر در همانندسازی DNA است. پلیمرازهای بسیاری هستند ولیpol η آنزیمی است که DNA تخریب شده با پرتوی فرابنفش را همانندسازی میکند. افراد با جهشهای این ژن دارای یک زیرمجموعهٔ XP، واریانتی از بیماری XP هستند.
تعمیر DNA و کاهش و افزایش خطر سرطان
بسیاری نشانگانهای سرطان به خاطر یک نقصان ارثی در توانایی تعمیر DNA است. هنگامی که یک جهش ارثی موجود در یک ژن تعمیر DNA باشد، که ژن تعمیر در یک شکل دگرشیافته بیان خواهند شد یا نخواهند شد. سپس کنش تعمیر شبیه کاهش خواهد بود و نتیجتاً DNA آسیب میبیند و گرایش به انباشتگی بر خود خواهد داشت. همانگونه که آسیبهای DNA میتواند خطاهایی را باعث شود، سنتز DNA نیز منجر به جهشهایی میگردد؛ که برخی از آنها میتواند برانگیزندهٔ سرطان باشند. جهشهای تعمیر DNA خطی-اصلی (Germ-line repair mutations) که خطر سرطان را افزایش میدهند در جدول زیر فهرست بندی شدهاند:
| ژن تعمیر DNA | پروتئین | مسیرهای تعمیری مؤثر | سرطانها با خطر افزایش یافته |
| گشادشدگی مویرگهای آتاکسی جهشیافته | ATM | جهشهای گوناگون در ATM کاهشHRR, SSA یاNHEJ | لوکمیا، لمفوما، سینه |
| نشانگان شکوفه | BLM (هلیکاز) | HRR | لوکمیا، لمفوما، روده بزرگ، سینه، پوست، شش، مجرای شنوایی، زبان، سرخنای، معده، لوزه، خشکنای، زهدان |
| سرطان سینه ۱ و ۲ | BRCA1 BRCA2 | HRR دو رشتهای میشکند و رشتهٔ دختری شکافدار میگردد | سینه، تخمدان |
| ژنهای کمخونی فانکونی
FANCA,B،C,D1,D2,E,F,G,I,J,L,M,N,O,P |
FANCA etc. | HRR و TLS | لوکمیا، تومورهای کبد، تومورهای جامد بسیاری مناطق |
| ژنهای سرطان رودهای-مقعدی غیرپولیپوزیس ارثی MSH2 MSH6 MLH1 PMS2 | MSH2 MSH6 MLH1 PMS2 | MMR | مشکلات رودهای-مقعدی، درونزهدانی، تخمدانی، معدی-رودهای (اشکالات معدی، روده کوچک، لوزالمعدی و صفراوی)، ادراری، مغزی (گلیبوبلاستوماها) و پوستی (کراتوآکاتوماها و آدنوماهای چرب) |
| ژن نشانگان لی-فراوْمنی TP53 | P53 | نقش مستقیم در HRR, BER, NER ایفا میکند و پاسخگوی تخریب DNA است این مسیرهای NHEJ وMMR برای این نشانگان هستند | سارکوماها، سرطانهای سینه، تومورهای مغزی، و کارسینوماهای فوق کلیوی |
| MRE11A | MRE11 | HRR و NHEJ | سینه |
| MUTYH | MUTYH گلوکوزیلاز | BER از یک جفت با 8-oxo-dG | سرطانهای رودهای-مقعدی، دوازدههای، تخمدانی، مثانهای و پوست |
| نشانگان شکست نیژمنگن | NBS (NBN) | NHEJ | سرطانهای لمفوئید |
| NTHL1 | NTHL1 | BER برای Tg, FapyG, 5-hC, 5-hU درdsDNA | سرطان روده بزرگ، سرطان درونزهدانی، سرطان دوازدههای، کارسینوما سلول پایهای |
| RECQL4 | RECQ4 | هلیکاز مشابهاً در HRR فعال است | کارسینومای سلول پایهای، کارسینومای سلول سنگفرشی، کارسینومای بینروپوستی |
| ژن نشانگان ورنرWRN | هلیکاز نشانگان ورنر وابسته به ATP | HRR, NHEJ, مسیر طولانی BER | سارکومای نرم بافتی، رودهای-مقعدی، پوست، تیروئید، لوزالمعده |
| ژنهای خشکپوستی رنگدانهدار XPA, XPB, XPD, XPF, XPG | XPA XPB XPD XPF XPG | رونویسی کامل NER رشتههای رونویسیشدهٔ ژنهای فعال را بهطور رونوشتبردار تعمیر میکنند | سرطان پوست (ملانوما و غیر-ملانوما) |
| ژنهای خشکپوستی رنگدانهدار XPC, XPE (DDB2) | XPC, XPE | ژنومیک جهانیNER، در هر دو DNAی رونویسیشده و رونویسینشده، آسیب میبیند. | سرطان پوست (ملانوما و غیر-ملانوما) |
| XPV (همچنین پلیمرازH نیز خوانده میشود) | DNAپلیمراز اِتا (Pol η) | سنتز ترا-آسیبش (TLS) | سرطان پوست (ملانوما، سلول سنگفرشی و پایهای) |