
| سندرم کرنس-سهیر | |
|---|---|
| تخصص |
چشمپزشکی |
| طبقهبندی و منابع بیرونی | |
| آیسیدی-۱۰ | H49.8 |
| آیسیدی-۹-سیام | 277.87 |
| اُمیم | ۵۳۰۰۰۰ |
| دادگان بیماریها | 7137 |
| ئیمدیسین | article/۹۵۰۸۹۷ |
| سمپ | D007625 |
| مرور ژن | |
سندرم کرنس-سهیر (انگلیسی: Kearns–Sayre syndrome) که به اختصار «KSS» نامیده میشود و با نامهایی همچون «اختلال آکیولوکرانیوسوماتیک» و «اختلال نوروماسکولار آکیولوکرانیوسوماتیک با فیبرهای قرمز خشن و راهراه» هم شناخته میشود، نوعی میوپاتی میتوکندریال است که معمولاً پیش از ۲۰ سالگی آغاز میشود. این بیماری در واقع فرم شدیدی از «اُفتالموپلژی خارجی مزمن پیشرونده» (به اختصار: CPEO) است که مشخصهاش، درگیری عضلات کنترلکننده حرکات «پلک» («لِوِیتور پالپبرا» و «اوربیکیولاریس آکیولای») و «چشم» (عضلات خارج چشمی) است. در نتیجهٔ این درگیریها، افتادگی پلک و فلج ماهیچههای چشمی رخ میدهد. در سندرم کرنس-سهیر، علاوه بر موارد یادشده، «رتینوپاتی رنگدانهای دوطرفه» و «بلوک قلبی» هم وجود دارد. سایر علائم احتمالی عبارتند از: «آتاکسی مخچهای»، «ناشنوایی»، «دیابت»، «کمبود هورمون رشد»، «کمکاری غده پاراتیروئید» و سایر اختلالات غدد درونریز و متابولیسم.
در هر دو بیماری فوق، گرفتاری عضلانی ممکن است در آغاز یکطرفه باشد، اما سرانجام دوطرفه خواهد شد و سیر آن نیز پیشرونده است.
علائم و نشانهها
بیماران مبتلا به سندرم کرنس-سهیر در آغاز همان علائم رایج در «اُفتالموپلژی خارجی مزمن پیشرونده» را پیدا میکنند و شروع علائم در دهههای اول و دوم زندگی است.
نخستین علامت، «افتادگی یکطرفهٔ پلک» یا «اشکال در بازکردن پلک» است که کمکم به سمت مقابل هم سرایت کرده و دوطرفه میشود. با پیشرفت این عارضه، کودک تلاش میکند گردن را به عقب برده و چانه را بالا بیاورد تا جلوی انسداد محور بینایی را بگیرد. در کنار این علامت، بهتدریج حرکاتِ کُره چشم هم محدود شده و فرد برای دیدن اطراف، مجبور است تا از چرخاندن گردن خود به اطراف یا بالا و پائین استفاده کند.
رتینوپاتی رنگدانهای

این بیماری موجب پیگمانتاسیون شبکیه، به ویژه در فاندوس خلفی چشم میشود. نمای ظاهری آن را به صورت «دانههای نمک و فلفل» توصیف کردهاند. در این حالت پیگمانتاسیونِ (رنگدانهسازیِ) گستردهٔ اپیتلیوم شبکیه رخ میدهد که بیشترین اثرش در «لکه زرد» مشاهده میشود. این موضوع برخلافِ بیماریِ «رتینیت پیگمنتوزا» است که رنگدانهسازی، بیشتر در بخشهای محیطیِ شبکیه رخ میدهد. نمای شبکیه در سندرم کرنس-سهیر مشابه آنچیزی است که در «دیستروفی میوتونیک نوع ۱» (DM1) هم دیده میشود. در این بیماران درجاتی از نابیناییِ شبهنگام (در نور کم) وجود دارد. کاهش حدت بینایی در این بیماران خفیف بوده و تنها در ۴۰ تا ۵۰ درصد بیماران مشاهده میشود.
اختلالات هدایت الکتریکی قلب
در بیشتر موارد، این اختلالات قلبی، سالها پس از وقوع افتادگی پلک و فلج ماهیچههای چشمی رخ میدهد. بلوک دهلیزیبطنی شایعترین نوعِ این اختلالات است و اغلب به سمت «بلوک درجه ۳ دهلیزیبطنی» (بلوک کامل هدایت الکتریکی از دهلیز به بطن) پیش میرود. علائم قلبیِ این عارضه شاملِ «سنکوپ»، «عدم توانایی در فعالیت جسمی» و «برادیکاردی» است.
سایر موارد
آنگونه که در نخستین توصیفهای این بیماری در سال ۱۹۶۵ میلادی و همچنین در سالهای آتی آمده بود، برخی دیگر از علائم کمنامتعارف آن عبارتند از: ضعف ماهیچههای صورت، ضعف ماهیچههای گلو، تنه و اندام، ناشنوایی، قد کوتاه، تغییرات الکتروانسفالیک (نوار مغزی)، آتاکسی مخچهای و افزایش سطح پروتئین در مایع مغزی-نخاعی
سببشناسی
در بیشتر بیماران، سندرم کرنس-سهیر بهطور خودبخود شروع میشود. در برخی دیگر از افراد نشان داده شده که بیماری، از طریق ارثی (اتوزومال غالب، اتوزومال مغلوب و میتوکندریال) ایجاد میشود. این بیماری هیچگونه ترجیح جنسی و نژادی نداشته و عوامل خطرزای شناختهشدهای هم ندارد. تا سال ۱۹۹۲ میلادی، تنها ۲۲۶ مورد اثباتشده از این بیماری در مقالات علمی گزارش شده بود.
ژنتیک
سندرم کرنس-سهیر در اثر پدیدهٔ حذف ژنی در دیانای میتوکندریایی (mtDNA) رخ میدهد. دیانای میتوکندریایی انحصاراً از مادر به ارث میرسد.این دیانای ۳۷ ژن دارد و به صورت یک کروموزوم حلقوی با ۱۶٬۵۶۹ جفتباز در میتوکندری قرار گرفتهاست. از این تعداد ژن، ۱۳ تای آنها مسئول ساخت پروتئینهای زنجیره انتقال الکترون هستند، ۲۲ تایشان آرانای حامل (tRNA) را میسازند و ۲ عدد هم در ساخت آرانای ریبوزومی (rRNA) شرکت دارند. آن ۱۳ ژنی که پروتئینهای زنجیره انتقال الکترون را کد میکنند، در فسفرگیری اکسایشی دخیل هستند. هرگونه جهش در این ۱۳ ژن، سبب میشود که چرخهٔ تولیدِ انرژی در میتوکندری مختل شود و بیشترین اثر آن هم در سلولهایی مشاهده میشود که شدیداً وابسته به «متابولیسم بیهوازی» هستند، همچون مغز، ماهیچههای اسکلتی، ماهیچههای قلبی، اندامکهای حسی و کلیه. درک این موضوع در شناخت علائم بیاریهای میتوکندریال مهم است.
بجز محل و شدت جهش ژنی، عوامل دیگری هم در بروز علائم اینگونه بیماریها دخیل هستند. میتوکندریها در جریان چرخهٔ سلولی، چه در دوران جنینی و چه در سالهای آتی، در حالِ تولید و بازسازی هستند. اما از آنجایی جهشهای ژنتیکی غالباً در دوران جنینی رخ میدهند، فقط سلولهایی دارای میتوکندی معیوب خواهند بود که در دوران جنینی، دچار جهش شده باشند و بقیه سلولها، میتوکندی سالم خواهند داشت. در نتیجه، گسترشِ میتوکندریهایِ معیوب در بدن و بافتهای مختل آن، یکدست نخواهد بود. به این پدیده «هتروپلاسمی (ناهمگونی)» میگویند که یک ویژگیِ مهمِ بیماریهای میتوکندریایی محسوب میشود. اینکه کدام عضو بدن، چه میزان میتوکندی معیوب دارد، بسته به این دارد که جهش ژنی در چه زمانی از حیات و در کدام بافت بدن رخ دادهاست. به همین دلیل است که دو فرد مبتلایی که عیناً یک جهش ژنی دارند، ممکن است علائم و نشانههای بسیار متفاوتی از هم داشته باشند. در مطالعهای که در سال ۱۹۹۲ میلادی توسط دکتر فیشل، دکتر قدسیان و همکاران انجام شد، دو بیمار گزارش شد که دقیقاً حذف ژنی مشابهی در ۴٬۹۷۷-bp داشتند، اما علائمشان بهکلی با یکدیگر متفاوت بود. یکی از آنها علائم کلاسیک سندرم کرنس-سهیر را داشت و آن دیگری علائمی کاملاً متفاوت داشت که امروزه به نام «سندرم پیرسون» شناخته میشود. آنچه که موضوع را پیچیدهتر میکند آن است که سندرم پیرسون در برخی موارد و با گذشت زمان، مبدل در سندرم کرنس-سهیر میشود.
پژوهشهای سالهای اخیر نشان دادهاست که پدیدهٔ دوپلیکاسیون دیانای میتوکندریایی هم نقش مهمی در علائم ظاهری بیمار دارد. این پدیده در سندرم کرنس-سهیر و سندرم پیرسون دیده میشود اما در «افتالموپلژی خارجی مزمن پیشرونده» وجود ندارد.
اندازهٔ حذف ژنی در سندرم کرنس-سهیر متغیر و در حدود «۱٫۳ تا ۸kb» است. محل آن هم در ژنوم میتوکندیال متفاوت است. شایعترین نوعِ حذفِ ژنی ۴٫۹kb است که از محل استقرار ۸۴۶۹ تا به محل ۱۳۱۴۷ رخ میدهد و در ۱/۳ بیمارانِ کرنس-سهیر دیده میشود.
تشخیص
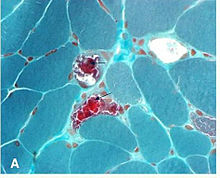
تشخیص این بیماری اغلب توسط «نوروافتالمولوژیستها» (متخصصان چشمی که فوقتخصص اعصاب چشمی دارند) انجام میشود. شک بالینی به این بیماری، باید بر اساس معاینات و یافتههای بالینی باشد. همچنین شک به میوپاتی باید در بیمارانی که فلج عصبیشان در اعصاب حرکتی چشمی، عصب ۴ و عصب ۶ مغزی است، افزایش یابد. معمولاً آزمایشهای تصویری اولیه، برای ردِ سایر بیماریهایِ شایعتر انجام میشود. تشخیص نهایی بر پایهٔ بیوپسی ماهیچهای و گاهی تأیید آن با «پیسیآر» برای جهشهای دیانای میتوکندریال است. البته لازم نیست بیوپسی از ماهیچهٔ چشمی خاصی انجام شود. فیبرهای عضلانی معمولاً با رنگ «گوموری تریکروم» رنگآمیزی میشود و زیر میکروسکوپ نوری مشاهده میشد. فیبرهایی که تعدادِ میتوکندیهای جهشیافتهاش زیاد باشد، تراکم میتوکندریال بالایی هم دارند و این موضوع باعث میشود که فیبرهای ماهیچهای در زیر میکروسکوپ، نمایی قرمزتیره و خشن داشته باشند که با آن «فیبرهای قرمز خشن» میگویند. گاهی از روشهای دیگری همچون رنگآمیزی آنزیمی میتوکندریال و آنالیزهای بیوشیمیایی بافت و مشاهده آنان در زیر میکروسکوپ الکترونی و همچنین آنالیز دیانای میتوکندریال ماهیچهها هم استفاده میشود.
مطالعات آزمایشگاهی
معمولاً در اثر فرایند افزایشِ متابولیسم بیهوازی و کاهش نسبت آدنوزین تریفسفات به آدنوزین دیفسفات، سطوح سرمی لاکتات و پیرووات افزایش مییابد. در مایع مغزی-نخاعی، سطح پروتئین به بیش از ۱۰۰ میلیگرم در دسیلیتر میرسد و سطح لاکتات آن نیز بالا میرود.
کنترل و درمان بیماری
سندرم کرنس-سهیر، در حال حاضر درمانِ قطعی ندارد. از آنجایی که این بیماری نادر است، تنها گزارشهای موردی دربارهٔ درمانهای انجامیافته موجود است و مشخص نیست که درجهٔ تأثیر این درمانها تا چه حد بودهاست. تاکنون چندین یافته و کشف امیدبخش گزارش شدهاست که ممکن است درهای جدیدی را برای درمان بیماری از طریق پژوهشهای بیشتر باز کند. به عنوان مثال، میتوکندیهای جهشیافته اغلب در سلولهای پیشساز ماهیچهها موسوم به «سلولهای سَتلایت» در بدن این بیماران دیده نمیشوند. در سال ۱۹۹۷ میلادی «شوبریج» و همکارانش پیشنهاد کردند که ممکن است با ترغیب «سلولهای سَتلایت» به ساختِ دوبارهٔ فیبرهای عضلانی در این بیماران، دوباره بتوان میتوکندریهایی با دیانای سالم به وجود آورد. در این پژوهش، فیبرهای بازسازی شدهٔ بیمار را در همان محل بیوپسی قرار دادند و مشاهده کردند که سلولهای ماهیچهای نسبت به میتوکندیهای اصلی (طبیعی) هوموپلاسمیک است. بدین ترتیب ممکن است در آینده و پیشرفت روشهای القای پرولیفراسیون و رشد «سلولهای سَتلایت» و بازسازی فیبرهای عضلانی، عملکرد و زندگی این بیماران به طرز مؤثری بهبود یابد.
در پژوهشی دیگر و در بیماری که سطوح پائین «کوآنزیم کیو ۱۰» سرمی داشت، تجویز ۱۲۰–۶۰ میلیگرم «کوآنزیم کیو ۱۰» به مدت ۳ ماه، سبب شد تا سطوح لاکتات و پیرووات خون به حالت عادی برگشته، بلوک درجهٔ ۱ قلبی بیمار بهبود یافته و حرکات چشمیاش نیز بهتر شود.
در تمامی بیمارانی که با علائم «افتالموپلژی خارجی مزمن پیشرونده» (CPEO) مراجعه میکنند، انجام ECG (نوار قلبی) توصیه میشود. در سندرم کرنس-سهیر و در صورت بروز اختلالات شدید هدایت الکتریکی قلب، کاشت «ضربانساز قلبی» عاقلانه است، حتی در بیمارانی که فاقد علائم قلبی هستند. غربالگری غدد درونریز هم باید انجام شود که میتوان از آن میان، به سنجش قند خون، تستهای تیروئیدی، کلسیم و منیزیم سرمی و سایر الکترولیتهای خون اشاره کرد. در ۳ درصد بیماران مبتلا به سندرم کرنس-سهیر، «هیپرآلدوسترونیسم» دیده میشود.
تاریخچه
علائم سهگانهٔ «افتالموپلژی خارجی مزمن پیشرونده» (CPEO)، رتینوپاتی رنگدانهای دوطرفه و اختلالات هدایت الکتریکی قلب، نخستین بار توسط «دکتر توماس پی. کرنس» (۲۰۱۱–۱۹۲۲) و «دکتر جرج پامهروی سهیر» (۱۹۹۲–۱۹۱۱) در دو بیمار در سال ۱۹۵۸ میلادی توصیف شد. یک مورد دیگر از بیماری که در نوجوان ۱۳ سالهای بود، در سال ۱۹۶۰ میلادی توسط «دکتر ب.و. یاخر» و همکاران گزارش شد. پیش از آن، نمونههای دیگری از «افتالموپلژی خارجی مزمن پیشرونده» که بهطور ناگهانی فوت میکردند، گزارش شده بود و معمولاً آنها را به دلیل «دیسریتمی قلبی» میدانستند. برخی گزارشها هم به پیگانتاسیون شبکیهای اشاره کرده بودند، اما هیچکدام این سهعلامت را بهطور همزمان باهم و در قالب یک سندرم، توصیف نکرده بودند. دکتر کرنس در سال ۱۹۶۵ میلادی گزارشی توصیفی از این بیماری و علائم سهگانهاش منتشر کرد. در سال ۱۹۸۸ میلادی نخستین ارتباط سندرم کرنس-سهیر و چند حذف ژنی در دیانای میتوکندریایی کشف شد. از آن پس، تعداد فراوانی حذف ژنی درارتباط این بیماری کشف و گزارش شدهاست.
- مشارکتکنندگان ویکیپدیا. «Kearns–Sayre syndrome». در دانشنامهٔ ویکیپدیای انگلیسی، بازبینیشده در ۱۸ دسامبر ۲۰۱۶.