
| بیماریهای ذخیرهای لیزوزومال | |
|---|---|
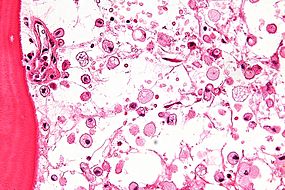 | |
| عکاسی ریزنگاری بیماری گوچر، با سلولهایی که دارای ویژگی مچالهشده سیتوپلاسم هستند. | |
| تخصص |
غدد درونریز و متابولیسم |
| طبقهبندی و منابع بیرونی | |
| آیسیدی-۱۰ | E75-E77 |
| سمپ | D016464 |
بیماریهای ذخیرهای لیزوزومال (به انگلیسی: Lysosomal storage disease) حدود پنجاه بیماری نادر ارثی هستند که اشکالات متابولیکی در عملکرد لیزوزومها (یک اندامک داخل سلولی) وجود دارد.
بیماری فابری با نقص در فعالیت یا کمبود آنزیم آلفا - گالاکتوزیداز (alpha-galactosidase A)، بیماری گوشه با نقص در آنزیم بتا-گلوکوسربروزیداز و بیماری نیمن پیک از این گروه هستند.
بیماریزایی
داخل همه سلولهای بدن اندامکی به نام لیزوزوم وجود دارد که توسط آنزیمهایی که درون آن وجود دارد مواد زائد و برخی مولکول های بزرگ موجود در سلول تجزیه شده و از بین می روند و یا برای استفاده مجدد و بازیافت، به اشکال ساده تر تبدیل می شوند. در این بیماریها به علت کمبود یکی از آنزیمهای لیزوزوم اختلالاتی در کارکرد سلول به وجود میآید که باعث تجمع ماده زائد ( لیپید، گلیکوپروتئین یا موکو پلی ساکارید) در بدن شده و به سلول آسیب رسانده و یا باعث مرگ سلول می شود. در این بیماری ها علائم پیشرونده، دائمی، در همه بیماران تقریباً یکسان و بی ارتباط با تغذیه و حوادث استرسزا میشود. این بیماری ها نادر هستند و معمولا به دلیل وقوع جهش های ژنتیکی اتفاق می افتند. نحوه وراثت این بیماری ها اغلب اتوزومال مغلوب است اما وراثت تعداد کمی از آن ها همچون بیماری فابری و سندرم هانتر به صورت وابسته به اکس مغلوب است. این بیماری ها عموما باعث مرگ کودکان در ماه ها و سال های ابتدایی زندگی می شوند.
طبقهبندی
به روشهای مختلف میتوان این بیماریها را طبقهبندی کرد مثلاً براساس نوع مادهای که متابولیزه نمیشود و در سلولها تجمع می یابد (طبقه بندی استاندارد)
الف. بیماری های ذخیرهای چربی
- اسفنگولیپیدوزها مانند بیماری گوشه و نیمن پیک
- گانگلیوزیدوزها مانند تای ساکس
ب. بیماریهای ذخیرهای موکوپلی ساکارید (موکوپلیساکاریدوزها) مانند سندرم هانتر و بیماری هورلر
ج. بیماری های ذخیرهای گلیکوپروتئین
طبقهبندی دیگر براساس نوع نقص که در این جدول نمایش داده شده است:
| نوع نقص پروتئینی | مثالهایی از بیماری | پروتئین گرفتار |
|---|---|---|
| نقص اولیه آنزیمهای لیزوزومی |
بیماری تای ساکس, I-cell disease, و اسفنگولیپیدوزها (مانند gangliosidosis ، بیماری گوشه و بیماری نیمن پیک) |
متنوع |
| پیرایش پسارونویسی آنزیمها | کمبود مالتیپل سولفاتاز | Multiple sulfatases |
| پروتئینهای ترابری غشاء | موکولیپیدوزها type II and IIIA | N-acetylglucosamine-1-phosphate transferase |
| Enzyme protecting proteins | گالاکتوسیالیدوز | Cathepsin A |
| Soluble nonenzymatic proteins | GM2-AP deficiency, variant AB, Niemann-Pick disease, type C2 | GM2-AP , NPC2 |
| Transmembrane proteins | SAP deficiency | Sphingolipid activator protein s |
| Niemann-Pick disease, type C1 | NPC1 | |
| بیماری ذخیره اسید سیالیک | Sialin | |
| Unless else specified in boxes, then ref is: | ||
جستارهای وابسته
- ویکیپدیای انگلیسی بیماریهای ذخیرهای لیزوزومال