| نویسنده(های) اصلی |
Chapman B, Chang J |
|---|---|
| انتشار ابتدایی | ۲۰۰۰ |
| انتشار پایدار | ۱٫۶۸
۲۶ اوت ۲۰۱۶ |
| مخزن | |
| نوشتهشده با | پایتون (زبان برنامهنویسی) و سی (زبان برنامهنویسی) |
| بنسازه رایانش | Cross-platform |
| گونه | بیوانفورماتیک |
| پروانه | Biopython License |
| وبگاه | |
بایو پایتون (به انگلیسی: Biopython) یک مجموعهٔ متن باز از ابزارهای غیر تجاری پایتون در زمینهٔ زیستشناسی محاسباتی و بیو انفورماتیک است. این مجموعه توسط جمعی از توسعه دهندگان با ملیتهای متفاوت توسعه یافتهاست.
این مجموعه شامل کلاسهایی برای نمایش فرایندهای زیستی و نمایش ریاضی این فرایند هاست. این مجموعه قادر به خواندن و نوشتن قالبهای متنوعی از فایل هاست. همچنین این مجموعه رابطی برای اتصال به پایگاههای داده ی زیستی برخط مانند پایگاههای NCBI را از طریق برنامهنویسی ارائه میکند. افزونههای متعددی، تواناییهای بایوپایتون را به مواردی مثل هم ترازی فرایند، ساختار پروتئین، ژنتیک جمعیت، تکامل نژادی، شماهای فرایند و یادگیری ماشین توسعه دادهاند. بیوپایتون یکی از پروژههای زیستی است که با هدف کاهش تکرار کد در زیستشناسی محاسباتی ایجاد شدهاست.
تاریخچه
توسعهٔ بایوپایتون در سال ۱۹۹۹ شروع شد و اولین نسخهٔ آن در ژوئیه ۲۰۰۰ منتشر گردید. در زمان توسعهٔ این مجموعه، مجموعههای مشابه دیگری نیز توسعه یافتند که تواناییهایی در زمینهٔ بیو انفورماتیک را به زبانهای برنامهنویسی شان افزودند؛ از آنها میتوان بایوپِرل (به انگلیسی: BioPerl)، بایورابی (به انگلیسی: BioRubby) و بایوجاوا (به انگلیسی: BioJava) را نام برد. اولین توسعه دهندگان این پروژه جف چانگ، اندرو دالک و برد چپمن بودند با این وجود تا امروز بیش از ۱۰۰ نفر در توسعه و نگهداری این پروژه مشارکت داشتهاند. در سال ۲۰۰۷ یک پروژهٔ مشابه پایتون به نام PyCogent نیز به ثبت رسید
حیطهٔ اولیهٔ بایوپایتون شامل دسترسی، نمایهگذاری و پردازش فایلهای فرایندهای زیستی است. با اینکه این موضوع هنوز هم یک هدف اصلی بایوپایتون است، طی چندین سال گذشته، افزونههای متعددی که برای آن عرضه شدهاند، حیطهٔ تواناییهای آن را برای پوشش مباحث زیستی دیگری افزایش دادهاند (بخش ویژگیهای کلیدی و نمونه را مشاهده کنید).
از نسخهٔ ۱٫۶۲ به بعد، بایوپایتون علاوه بر پایتون ۲ اجرا بر روی پایتون ۳ را نیز پشتیبانی میکند.
طراحی
هر جا که ممکن بوده بایو پایتون از قالب و روش زبان برنامهسازی پایتون برای تعامل با کاربر استفاده کردهاست تا استفاده از آن برای کاربران آشنا به این زبان برنامهسازی آشنا باشد. به عنوان مثال اشیای Seq و SeqRecord را میتوان با تکه کردن آنها و دقیقاً مثل stringها و listهای پایتون تغییر داد. این پروژه همچنین طوری طراحی شده که به لحاظ عملکرد مشابه سایر پروژههای زیستی مثل BioPerl باشد.
بایوپایتون قادر است که بیشتر قالبهای فایل رایج را برای هر کدام از زمینههای کاری اش بخواند و بنویسد. جواز این مجموعه با بسیاری از جوازهای دیگر نرمافزارها سازگار است که به بایوپایتون این امکان را میدهد که در طیف وسیعی از پروژههای نرمافزاری مورد استفاده قرار گیرد.
ویژگیهای کلیدی و نمونه
توالیها
یک مفهوم کلیدی در بایو پایتون، توالی زیستی است که توسط کلاس Seq نمایش داده میشود. یک شی از نوع Seq در بایوپایتون، از بسیاری جوانب به یک رشته در زبان پایتون شبیه است. این کلاس از نشانهگذاری پیشفرض پایتون برای برش رشتهها پشتیبانی میکند، قابل الحاق به سایر دنباله هاست و همچنین تغییرناپذیر (به انگلیسی: immutable) است. به علاوه این کلاس شامل رویههایی مخصوص توالیها نیز هست و الفبای زیستی خاص مورد استفاده را مشخص میکند.
>>> # This script creates a DNA sequence and performs some typical manipulations
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import IUPAC
>>> dna_sequence = Seq('AGGCTTCTCGTA', IUPAC.unambiguous_dna)
>>> dna_sequence
Seq('AGGCTTCTCGTA', IUPACUnambiguousDNA())
>>> dna_sequence[2:7]
Seq('GCTTC', IUPACUnambiguousDNA())
>>> dna_sequence.reverse_complement()
Seq('TACGAGAAGCCT', IUPACUnambiguousDNA())
>>> rna_sequence = dna_sequence.transcribe()
>>> rna_sequence
Seq('AGGCUUCUCGUA', IUPACUnambiguousRNA())
>>> rna_sequence.translate()
Seq('RLLV', IUPACProtein())
نشانهگذاری توالی
کلاس SeqRecord توالیها را توصیف میکند و همچنین اطلاعاتی از آنها را در قالب اشیایی از جنس SeqFeature ارائه میدهد. مثل نام توالی، توضیحات آن و ویژگیهای آن.
هر شی SeqFeature نوع و مکان یک ویژگی توالی را مشخص میکند. از انواع مختلف یک شی SeqFeature میتوان 'gene'، 'CDS'، ‘repeat_region’ و ‘mobile_element’ را نام برد. موقعیت ویژگیها در یک توالی میتواند دقیق یا تقریبی باشد.
>>> # This script loads an annotated sequence from file and views some of its contents.
>>> from Bio import SeqIO
>>> seq_record = SeqIO.read('pTC2.gb', 'genbank')
>>> seq_record.name
'NC_019375'
>>> seq_record.description
'Providencia stuartii plasmid pTC2, complete sequence.'
>>> seq_record.features[14]
SeqFeature(FeatureLocation(ExactPosition(4516), ExactPosition(5336), strand=1), type='mobile_element')
>>> seq_record.seq
Seq('GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC...GCC', IUPACAmbiguousDNA())
ورودی و خروجی
بایوپایتون میتواند برای دریافت و تحویل اطلاعات با تعدادی از فرمتهای رایج دنباله کار کند. از این دنبالهها میتوان FASTA، FASTQ، GenBank، Clustal، PHYLIP و NEXUS را نام برد. در هنگام خواندن فایلها، اطلاعات توصیف شده در آنها برای مقدار دهی به اعضای کلاسهای بایوپایتون مانند SeqRecord مورد استفاده قرار میگیرد. این عملیات واسطی برای تبدیل اطلاعات ذخیره شده در یک قالب فایل به قالب دیگر را مهیا میکند.
ممکن است حافظهٔ یک سیستم رایانهای نوعی برای بارگذاری فایلهای توالی بسیار بزرگ کافی نباشد. برای حل این مشکل، بایوپایتون، گزینههای متنوعی برای دسترسی به اطلاعات فایلهای حجیم ارائه میکند. فایلها میتوانند بهطور کامل و توسط ساختارهای دادهٔ زبان پایتون مثل لیستها و آرایههای انجمنی در حافظه بارگیری شوند و دسترسی به اطلاعات به قیمت افزایش مصرف حافظه سریع تر شود؛ یا به عنوان روشی جایگزین، اطلاعات در مواقع نیاز از دیسک خوانده شوند که منجر به دسترسی کند به اطلاعات خواهد شد ولی منابع حافظهٔ کمتری مصرف خواهد کرد.
>>> # This script loads a file containing multiple sequences and saves each one in a different format.
>>> from Bio import SeqIO
>>> genomes = SeqIO.parse('salmonella.gb', 'genbank')
>>> for genome in genomes:
... SeqIO.write(genome, genome.id + '.fasta', 'fasta')
دسترسی به پایگاههای دادهٔ برخط
کاربران بایوپایتون میتوانند از طریق افزونهٔ Bio.Entrez به بارگیری اطلاعات از پایگاههای دادهٔ بر خط NCBI بپردازند. تمام رویههای فراهم شده توسط موتور جستجوی Entrez نیز از طریق رویههایی معادل در این افزونه قابل دسترسی هستند. مثلاً جستجو برای اطلاعی خاص و بارگیری آن.
>>> # This script downloads genomes from the NCBI Nucleotide database and saves them in a FASTA file.
>>> from Bio import Entrez
>>> from Bio import SeqIO
>>> output_file = open('all_records.fasta', "w")
>>> Entrez.email = 'my_email[at]example.com'
>>> records_to_download = ['FO834906.1', 'FO203501.1']
>>> for record_id in records_to_download:
... handle = Entrez.efetch(db='nucleotide', id=record_id, rettype='gb')
... seqRecord = SeqIO.read(handle, format='gb')
... handle.close()
... output_file.write(seqRecord.format('fasta'))
تکامل نژادی

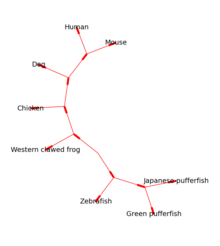
افزونهٔ Bio.Phylo ابزارهایی برای کار کردن و نمایش گرافیکی درختهای تکامل ژنتیکی را ارائه میکند. تعداد زیادی از قالبهای فایل برای خواندن و نوشتن مورد استفاده قرار میگیرند
درختهای ریشه دار را میتوان توسط اعضای ASCII یا با استفاده از رویههای matplotlib (شکل ۱ را ببینید) ساخت و درختهای بدون ریشه توسط Graphviz قابل ساخت هستند (شکل ۲ را ببینید).
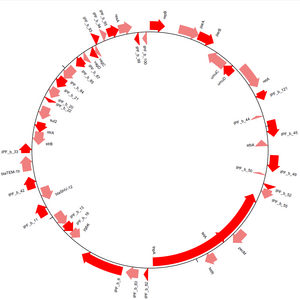
نمودارهای ژنوم
افزونهٔ GenomeDiagram رویههایی برای نمایش گرافیکی توالیها در بایوپایتون را ارائه میدهد. نمودارها میتوانند به صورت
دایرهای، خطی و … و با انواع مختلفی از قالبهای خروجی ساخته شوند. مثلاً PDF و PNG. (شکل ۳ را ببینید)